
Diagnosis, Targeted therapies
Conventional cancer drugs are cellular poisons that block replication or some other aspect of cell growth. These drugs affect all cells – healthy or cancerous.
Conventional cancer drugs are cellular poisons that block replication or some other aspect of cell growth. These drugs affect all cells – healthy or cancerous – causing debilitating side effects. New drugs are now being "rationally" designed to knock out or inactivate specific molecules in a cancer-causing pathway. These "targeted therapies" can efficiently attack cancer cells with greatly reduced side effects. Explore this section to learn how these therapies work by blocking receptors, targeting activators, or attacking tumors. Blocking Receptors In this section, hear what experts have to say about drugs that disrupt the function of receptors on a cell's surface. Breast cancer – the most prevalent cancer affecting women – is being successfully treated using these "receptor blockers." Click forward to find out more about two breast cancer drugs and their receptor targets. Larry Norton, M.D., Memorial Sloan-Kettering Cancer Center: “Well many cancers actually require hormones to grow. Breast cancer is a very good example. Breast cancer arises in the breast, and the breast is an organ that is responsive to the female hormone, estrogen.†“Cancers derived in the breast, therefore, especially in older women, tend to require estrogen to grow. If we can starve that cell of estrogen, we can actually make that cancer cell die.†“And that’s what we do right now with a couple of different approaches. One of the drugs is called tamoxifen, which actually attaches to the estrogen receptor.†“The estrogen receptor is a protein that is found in many breast cancer cells that finds estrogen in surrounding blood, takes it into the cell, and signals the cell to grow.†“Well, tamoxifen attaches to the estrogen receptor so the estrogen can’t attach to the receptor. And when tamoxifen and the receptor for estrogen go into the nucleus of the cell, it attaches to the DNA and actually signals the cell not only to grow but to die. So that’s a very important molecule.†“One of the really important things to know about cancer is that no two cancers are the same. They're all different. There is tremendous variations in the molecules that are involved in making a cell cancerous. By identifying those molecules in the individual case, we can individualize therapy – give people the medicines they need, and not give medicines to people if they are not going to benefit.†The estrogen receptor is not the only target in the treatment of breast cancer. Another important target is the human epidermal growth factor receptor (Her-2), which is overexpressed in 25% of breast cancers, leading to cellular growth and proliferation. “If a cell has too much her2, the cell will be dividing too often because the cell will be interpreting many stimuli in the environment as stimuli cause it to divide whereas a normal cell wouldn’t. It makes that cell have too much of a tendency for cell division and to go on to form a lump and to go on and spread into the surrounding tissue like an invasion or to grow on other parts of the body.†“We've developed an antibody that attaches to her2. Antibodies are the proteins that your body makes naturally in response to infections. And your body normally makes lots of antibodies, and should be that's how come you fight lots of infections. However, we can now make her2 antibodies – we call it Trastuzumab or Herceptin – outside the human body and give it to the patient intravenously if that patient has a cancer, particularly a breast cancer, with too much her2 in it.†“Antibody flows in the blood, finds the cancer cell, attaches to the her2, and inactivates it so now it can’t act as a molecule, the cell is not getting the stimulus that is giving it information to make it divide so it stops dividing and stops dividing it often goes on to kill itself, called programmed cell death.†Herceptin is one example of a treatment that targets a specific molecule in a particular type of cancer. As more of these precisely targeted therapies are developed, it will become increasingly important to understand which molecules play important roles in a particular individual’s cancer. See the Pharmacogenetics section for more information on patient-specific treatment. Targeting activators Tyrosine kinases are a family of activator proteins that trigger the cell signaling process leading to cell growth and division. These kinases are often mutated in cancer. Several anti-cancer drugs have been developed to block specific members of this protein family, including Iressa™ and Gleevec™. Click the forward arrow to find out more about how these drugs work in treating cancer. Charles Sawyer, M.D. is professor of medicine and director of the Prostate Cancer Program Area at U.C.L.A. Johnsson Comprehensive Cancer Center, and an investigator of the Howard Hughes Medical Institute. He works on therapies that target specific mutations in prostate cancer and chronic myeloid leukemia (CML). Here he describes his work developing a targeted therapy for CML patients with resistance to the anti-cancer drug Gleevec. “CML stands for chronic myeloid leukemia, which is a blood cancer. It is different from many cancers because it starts very slowly and patients when they're first diagnosed don't have many symptoms. They just have a high white blood cell count that is detected by their physician when they get a routine check up. The incidence of CML is about 5,000 new cases a year in the U.S., another 5,000 in Europe, so, 10,000 patients a year. Patients tend to have CML for five or six years and then, and it's easily controlled with oral chemotherapy drugs until it turns into an aggressive very acute leukemia called blast crisis. And then it becomes a fatal illness. CML is caused by a chromosome translocation known as the Philadelphia chromosome, which occurs in a stem cell in the bone marrow. Presumably a single cell develops that and over a period of years that cell gets a growth advantage and eventually results in a leukemia. The translocation involves two genes. The main gene is a tyrosine kinase called Abl. And the fusion of BCR from one chromosome to Abl on the other creates a kinase that's constantly on. And that enzyme causes this entire disease. We know that by putting this enzyme into a mouse model. You get essential CML in a mouse.†“So Gleevec is a pill taken once a day and it is, works remarkably well in all phases of CML. So at the beginning, it was tested in patients with CML who had it for many years and failed standard chemotherapy type treatments. It worked extremely well there, and now is used as front line therapy in essentially all patients with CML. It's taken once a day, there's very little in the way of side effects. Patients live essentially a normal life. If patients are in the later stages' of CML when they start Gleevec, it works quite well, but resistance develops generally after six months to a year. When patients who are newly diagnosed with CML start Gleevec, so far it appears to be working remarkably well for three to four years now of follow-up. That's the longest follow-up we have. But about 15% of patients have developed resistance in that early diagnosis group.†“That resistance for the most part is due to the outgrowth of subclones of the CML that have mutations in the Abl kinase domain, right at the places where the Gleevec drug binds. And there are a number of different mutations that have been described and collectively they all share in common the property of interfering with the ability of Gleevec to bind tightly to Abl.†“They come about most likely because in the process of growing, the CML clone makes mistakes in DNA replication and generates a diverse repertoire of mutations – most of which are probably irrelevant and just disappear but under the face of selective pressure of a drug, you get outgrowths. They get a growth advantage and they grow up. What's I think is clear is the drug itself is not causing mutations; it's not mutagenic. And when we look using very sensitive tools, it looks like the mutations are already there in most patients and so the die might be cast early on in the disease and it speaks to the need of having perhaps a cocktail of drugs to combat this much like the treatments for HIV virus is based. Well, I can give a number but the natural history of CML is actually a little different now in 2004 than it was four years ago because essentially everyone is now on Gleevec. But if you start on Gleevec in the late stage of the disease, 100% of patients have a resistance within about a year or two. If you start Gleevec at the beginning of the disease, about 15% have resistance within three years. We don't know beyond that what will happen but the curve seems to be continually dropping down gradually but there's no evidence that anyone's been cured with Gleevec and when we use PCR to look for residual CML cells in patients, even those that are doing fantastic, we still see residual cells by PCR. So, most of us believe that there's a reservoir of CML cells left escaping Gleevec, not necessarily expanding but just sitting there. And whether or not that reservoir will be a big problem in another five years of follow-up, we'll just have to see.†“So when we had this collection of mutations that were staring at us in the face, we first wanted to understand why did they cause resistance and the reason is they interfere with the ability of the Abl kinase to actually get in the right shape to bind Gleevec. One of the unique properties of Gleevec is that it locks Abl into a certain off configuration, turning the enzyme off.†“The kinase has to be pretty flexible to get into that shape. And most of these mutations occur in regions that interfere with this flexibility. So that led to the idea that maybe an inhibitor that would overcome resistance, would in a sense have to be a little sloppier. Maybe not put so many rules on the shape of the Abl kinase.†“After screening through a number of these and talking about this, I was contacted by Bristol-Myers Squibb who had such a compound that was essentially ready for clinical development. It had all the right pharmaceutical properties of a drug rather than just a lab reagent that was tested in mice and so forth and remarkably was effective against all of the Gleevec resistant mutants that we know of, except for one.†“So, now patients are on this compound. It's in phase one testing it for patients with CML who have Gleevec resistance in essentially almost all of these patients have these mutations and it looks quite promising in these clinical trials.†“I think in a more general way, this is a paradigm for how we will do this with the whole range of kinase inhibitors that are not quite as fully developed as the Gleevec story, but are coming along. I think the next example will be lung cancer, for which the EGF receptor happens to be the driver in at least 10% of lung cancer patients in the U.S. due to a mutation that was just recently described. In phase one study. The lung cancer patients in that study, a few of them had some benefit. That led to the decision empirically now to do a large study in lung cancer. And when that was done, about 10 percent of patients had miraculous types of responses. And the in other 90 percent nothing happened so that raises some conundrums for clinical development is obviously you would like to understand what was it about those 10 percent of patients.†Iressa targets a tyrosine kinase activator called epidermal growth factor receptor (EGFR). One half of this protein is a receptor on the surface of the cell that binds to signaling molecules. The other half of the protein activates additional proteins inside the cell to trigger cell growth and division. Two things have to happen before EGFR can trigger cell growth and division: a signaling molecule has to bind to the receptor part of EGFR, and a small molecule ATP has to bind to the activator part of EGFR. ATP fuels the reaction. Iressa is a drug that binds to the same part of EGFR as ATP. It prevents ATP from binding, and blocks the growth signal from being sent to the nucleus. Iressa does not work for everyone with lung cancer. In order to understand this, scientists have looked at the sequence of the gene that encodes EGFR in patients that respond to the drug and patients that do not. The patients who respond well to Iressa all have mutations in the ATP-binding region of the EGFR protein. These mutations appear to make the protein bind ATP more tightly, increasing the amount of signal that is passed on to the nucleus. These mutations also appear to make the protein bind Iressa more tightly. It is not yet clear why this makes such a difference in a patient’s response to Iressa. It may be that Iressa only affects tumors that are caused by a mutation in EGFR. If so, Iressa would be analogous to Gleevec, a drug that targets the mutant activator protein Bcr-Abl.
female hormone estrogen, breast cancer cells, breast cancer drugs, sloan kettering cancer center, sloan kettering cancer, memorial sloan kettering, memorial sloan kettering cancer center, larry norton, kettering cancer center, breast cancer, therapies work, receptor blockers, new drugs, estrogen receptor, tamoxifen, receptors, poisons, older women, cancers
- ID: 1010
- Source: DNALC.IC
Related Content

1011. Diagnosis, Targeted therapies: Blocking receptors
In this section, hear what experts have to say about drugs that disrupt the function of receptors on a cell's surface.

16839. Gallery 40: Harold Varmus
Harold Varmus, President and Chief Executive Officer of Memorial Sloan-Kettering Cancer Center.

16995. New York Stories: Restriction Enzyme Analysis
New York high school students interview Dr. Scott Lowe of Memorial Sloan-Kettering Cancer Center about using restriction enzyme analysis in cancer research, then perform the experiment.

16853. Biography 40: Harold Eliot Varmus (1939 -)
Harold Varmus and Mike Bishop worked out how retroviruses transform normal cells to cancerous ones.

1016. Diagnosis, Targeted therapies: Targeting activators, Sawyer 4
Professor Charles Sawyer explains that EGF receptor happens to be the driver in at least 10% of lung cancer patients in the U.S.
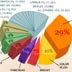
956. Causes, Smoking: Lung cancer epidemic
This section covers the smoking epidemic in the U.S. and the 163,000 Americans that die each year from lung cancer, which is greater than deaths caused by prostate, breast, colon, and pancrease cancers combined.

15713. Denise
Genetic testing changed Denise's life.

15938. What is breast cancer?
BRCA1, on chromosome 17, is one of the genes associated with hereditary breast cancer.

15939. What is breast cancer?
BRCA2, on chromosome 13, is one of the genes associated with hereditary breast cancer

15079. Family affliction, Denise
Denise talks about her family's affliction with inherited breast cancer and her decision to have her breasts removed as a preventive measure.