
Hotspot for Autism Genes
Several lines of research are converging to show how opposing genetic pathways can lead to autism.
Autism, a spectrum of often heartbreaking behavioral traits, also can be studied as a mystifying collection of genetic variants. Several lines of research are converging to show how opposing genetic pathways can lead to autism — perhaps laying important groundwork in understanding the biology of the disorder. “We’d all like to move in a straight line from the gene to the brain changes to the behavior,†says Ed Cook, an autism researcher at the University of Chicago. “But there are many steps in between.†Intriguing new clues are showing what some of these steps might be. One of the more frequent genetic anomalies is found on chromosome 15. Normally a person inherits one set of chromosomes from each parent, but in 1 to 3 percent of patients with autism, extra copies of a particular stretch of DNA are inherited from the mother but only one from the father. The double or triple dose of this region, designated 15q11-q13, is noteworthy because the same portion of maternal DNA is absent in most cases of a related condition, Angelman syndrome. Patients with this latter syndrome show mental retardation and signs that can be diagnosed as autism, including absence of speech, epilepsy, and movement difficulties. Despite seemingly opposite effects on this chromosomal region, both circumstances can lead to autism. One explanation may lie in a gene called UBE3A, a so-called imprinted gene, meaning that the paternal or maternal copy is expressed in varying proportions. Normally UBE3A is expressed in a maternal-specific manner, particularly in the brain; the paternal copy is “silenced†or inactivated. The absence of this gene from the maternal chromosome is a cause of Angelman syndrome. This maternal-specific phenomenon, along with another gene that may be involved, holds clues to autism and related disorders. The second gene, termed MECP2, mutates in a third neurological condition, Rett syndrome, which overlaps with autism in several ways: delays in development, language impairment, and repetitive movements. MECP2 is located not on chromosome 15 but on the X chromosome. Thought to play a role in silencing other genes, MECP2 is believed to act at an important control region near UBE3A to help regulate the gene. To test whether boosting levels of MECP2 would be a therapeutic option for Rett syndrome, Huda Zoghbi and colleagues at Baylor College of Medicine, Houston, developed a line of transgenic mice that express higher than normal amounts of the gene’s protein product, MeCP2. Their findings, published online Sept. 6 in Human Molecular Genetics, have implications for autism as well. After a period of normal development—consistent with both Rett syndrome and some cases of autism—the transgenic mice began to show signs of neurological disorder, including seizures and paw-clasping; many stopped grooming themselves and died prematurely. The authors do not draw overt parallels between mice overexpressing MECP2 and human patients with autism. However, Janine LaSalle and colleagues at the University of California, Davis, medical school have found increased levels of MECP2 in a few patients with autism or autism-like disorders—suggesting that excessive amounts of MECP2 may be a factor in autism. “Our findings unequivocally show that doubling MECP2 levels affects brain development and plasticity, making duplications of this gene a potential mechanism in some cases of autistic disorders,†says Zoghbi. UBE3A complicates the picture. LaSalle and colleagues have shown that mice lacking MECP2 have low levels of UBE3A. Because absence of UBE3A is a hallmark of Angelman syndrome, the research again seems to conflict: too much or too little MECP2 results in two distinct disorders, both of which share features with autism. The team’s report, published online Dec. 22 in Human Molecular Genetics, shows how apparently contradictory biochemical pathways may converge to produce the syndrome of autism. Studying brain tissue taken after death from patients with Rett syndrome, Angelman syndrome, and autism, the authors found reduced levels of both UBE3A and MECP2, mirroring the findings with the transgenic mice. They also found reductions in another gene that may be one missing piece of the puzzle. Located in the suspect region of 15q11-q13, this gene, GABRB3, encodes for part of a receptor for gamma-aminobutyric acid (GABA), a neurotransmitter that slows brain cell activity. According to James Sutcliffe, a genetics researcher at Vanderbilt University, “It’s possible that overexpression not only of UBE3A but of GABRB3 contributes to autism in people with extra copies of 15q11q13.†On the other hand, Sutcliffe surmises, abnormal imprinting control in this region (presumably involving MECP2) may explain the reduced expression of these genes in Rett syndrome and some cases of autism. “It’s an exciting finding,†Sutcliffe says, noting that linkage studies— which look for culprit genes in families with autism—point to the region of 15q11-q13 containing GABRB3 and two other genes that function in GABA receptors. As the roles of these and many other genes become better understood, scientists will develop a clearer picture of what happens in the brain to produce autism and related disorders. Zoghbi hypothesizes that mutations in these genes disrupt plasticity (the process through which brain cells make connections), strengthening or weakening the connections depending on specific experiences. Incremental gains can and do lead to new treatments: clinical trials are under way to see whether a drug to enhance synaptic strength can improve memory, language, and behavior in adults with Fragile X syndrome, an inherited form of mental retardation. Although autism research has not yet reached this level, work on the foundation has begun.
autism, asd, autisitic, gene, genetic, inherited, q13, chromosome, 15, UBE3A, MECP2, GABRB3, 15q11q13, fragile x,
- ID: 881
- Source: DNALC.G2C
Related Content
471. Chromosome Map of Disorders and Processes
An interactive chromosome map of the genes and loci associated with cognitive processes and disorders.
908. Autism Candidate Genes
Use this chromosome map to explore genes associated with autism.

916. GABRB3 Gene
GABA is the main inhibitory neurotransmitter in the adult brain. GABRA3 is a candidate gene for autism.

917. UBE3A Gene
Mutations to the UBE3A gene can cause Angelman syndrome. It is also a candidate gene for autism.

15945. What is Fragile X?
The FMR1 gene produces a protein involved in making cellular connections in the brain.

2364. Fragile X Syndrome - Inheritance
Doctor Gul Dolen explains that Fragile X syndrome is not a mendelian disorder, because the inheritance pattern in slightly different.
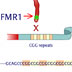
15946. What causes Fragile X?
The FMR1 gene produces a protein involved in making cellular connections in the brain. If this gene carries many repeats of the nucleotides CGG at one end, it is deactivated. People with this mutation display mental impairments or retardation. Fragile X s

2367. Biochemical Treatments for Autism?
Doctor Gul Dolen discusses how new biochemical treatments for Fragile X Syndrome may be used to treat autism.

2363. Fragile X Syndrome
Doctor Gul Dolen describes the key characteristics of Fragile X syndrome, which can include problems with language, mental retardation, and symptoms of autism.

2368. MGluRs and Treating Different Autisms?
Doctor Gul Dolen explains that there are many single-gene disorders on the autism spectrum, which may or may not respond to mGluR-related treatment.